
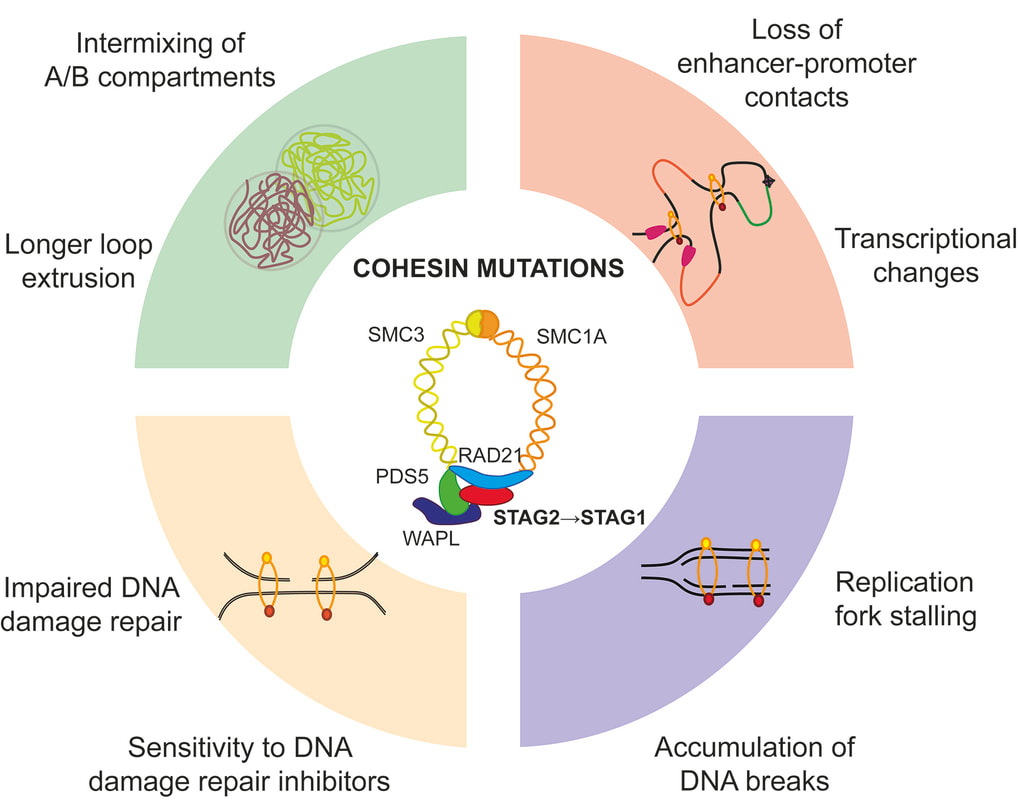
(Jann and Tothova, Blood 2021)
Our Research
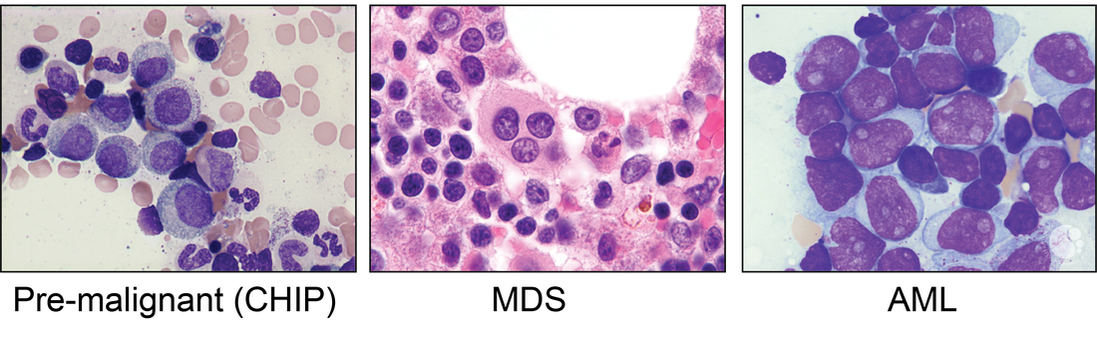

The primary focus of the Tothova laboratory is investigation of the biology, genetics and treatment of myeloid malignancies, including the premalignant state of clonal hematopoiesis (CHIP), myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). Our goal is to contribute to our understanding of the effect of chromatin organization on hematopoietic stem cell (HSC) transformation in the context of mutations in cohesin genes and other epigenetic modulators recurrently mutated in myeloid malignancies. We employ a combination of genomic, genome engineering, mouse modeling, single cell sequencing as well as classical biochemistry, cellular and molecular biology approaches to answer disease-relevant questions with the goal to identify novel therapeutic targets that can be translated to true patient benefit in the future.
The key questions that guide are work are understanding (1) how cohesin mutations promote clonal dominance and disease progression; and (2) how we can selectively target cohesin-mutant cells and intercept disease progression. Below are some of the currently ongoing projects:
1, How does the chromatin structure change during the progression of cohesin mutant myeloid disease and can we target it to avert progression to cancer?
Cohesin complex is an essential multimeric protein complex that forms a ring structure around the DNA and is involved in a number of basic cellular functions, including sister chromatid cohesion, chromatin organization and transcriptional control (Jann and Tothova, Blood 2021). Recurrent loss of function mutations in the cohesin genes STAG2, SMC3, SMC1A and RAD21 have been found in MDS, AML, bladder cancer, breast cancer, glioblastoma and Ewing sarcoma. We have developed CRISPR/Cas9-based engineering methods to target primary human and mouse hematopoietic stem cells to model different stages of myeloid malignancy in order to address this question (Tothova et al, JCI Insight 2021), and have access to different chromatin topology mapping studies (Hi-C, Micro-C) and a rich collection of patient samples to validate our findings.
2, What is the role of innate immune activation and inflammation during cohesin-mutant MDS development?
During the process of cancer development, cancer cells acquire genetic and epigenetic changes that promote a number of different characteristics that help them win over healthy cells. Some of these changes include impaired differentiation, increased proliferation and pro-survival properties, and resistance to inflammatory stress. In addition, cancer cells can also acquire the ability to evade the immune system. We have recently identified CTCF and multiple cohesin genes as key suppressors of PD-L1 and important regulators of immune evasion but also interferon signaling (Oreskovic et al, PNAS 2022). We are now pursuing studies aimed to understand the source of innate immune activation and the competitive advantage that cohesin mutations confer mutant hematopoietic stem and progenitor cells during the process of CHIP to MDS progression.
3, The role of transcription deregulation and splicing in cohesin-mutant disease
Mutations in the cohesin complex and splicing factor SF3B1 are frequent genetic drivers in patients with MDS and AML and they tend not to co-occur in the same patient. We have recently discovered that cohesin-mutant cells are exquisitely sensitive to splicing modulators, and that these can sensitize cells to additional PARP inhibition and/or chemotherapy (Wheeler, Martin et al, Science Translational Medicine 2024). We are now pursuing studies to dissect the biochemical interaction between the cohesin complex and the SF3B splicing complex and investigate how these two complexes may participate in non-canonical functions in splicing and chromatin organization.
4, Cohesin as regulator of R loop formation.
Cohesin-mutant cells accumulate DNA damage and have deficient DNA damage repair pathways and we have previously described the contribution of replication fork stalling to this phenotype (Tothova et al, JCI Insight 2021). Ongoing work in the lab is focused to understand the contribution of transcriptional stress in the form of R loop formation, and our preliminary data suggest that the cohesin complex is an important regulator of R loop formation.
5, Cohesin mutations as progression lesions in RUNX1 familial platelet disorder
Cohesin mutations are important progression lesions in a number of familial MDS/AML predisposition syndromes, including the RUNX1 familial platelet disorder (FPD), Trisomy 21 Down syndrome-associated acute megakaryoblastic leukemia (AMKL) and GATA 2 deficiency syndrome. We are interested in understanding the mechanisms by which cohesin mutations are selected for during MDS progression in these germline disorders and identifying ways to intercept progression to MDS and AML.
The key questions that guide are work are understanding (1) how cohesin mutations promote clonal dominance and disease progression; and (2) how we can selectively target cohesin-mutant cells and intercept disease progression. Below are some of the currently ongoing projects:
1, How does the chromatin structure change during the progression of cohesin mutant myeloid disease and can we target it to avert progression to cancer?
Cohesin complex is an essential multimeric protein complex that forms a ring structure around the DNA and is involved in a number of basic cellular functions, including sister chromatid cohesion, chromatin organization and transcriptional control (Jann and Tothova, Blood 2021). Recurrent loss of function mutations in the cohesin genes STAG2, SMC3, SMC1A and RAD21 have been found in MDS, AML, bladder cancer, breast cancer, glioblastoma and Ewing sarcoma. We have developed CRISPR/Cas9-based engineering methods to target primary human and mouse hematopoietic stem cells to model different stages of myeloid malignancy in order to address this question (Tothova et al, JCI Insight 2021), and have access to different chromatin topology mapping studies (Hi-C, Micro-C) and a rich collection of patient samples to validate our findings.
2, What is the role of innate immune activation and inflammation during cohesin-mutant MDS development?
During the process of cancer development, cancer cells acquire genetic and epigenetic changes that promote a number of different characteristics that help them win over healthy cells. Some of these changes include impaired differentiation, increased proliferation and pro-survival properties, and resistance to inflammatory stress. In addition, cancer cells can also acquire the ability to evade the immune system. We have recently identified CTCF and multiple cohesin genes as key suppressors of PD-L1 and important regulators of immune evasion but also interferon signaling (Oreskovic et al, PNAS 2022). We are now pursuing studies aimed to understand the source of innate immune activation and the competitive advantage that cohesin mutations confer mutant hematopoietic stem and progenitor cells during the process of CHIP to MDS progression.
3, The role of transcription deregulation and splicing in cohesin-mutant disease
Mutations in the cohesin complex and splicing factor SF3B1 are frequent genetic drivers in patients with MDS and AML and they tend not to co-occur in the same patient. We have recently discovered that cohesin-mutant cells are exquisitely sensitive to splicing modulators, and that these can sensitize cells to additional PARP inhibition and/or chemotherapy (Wheeler, Martin et al, Science Translational Medicine 2024). We are now pursuing studies to dissect the biochemical interaction between the cohesin complex and the SF3B splicing complex and investigate how these two complexes may participate in non-canonical functions in splicing and chromatin organization.
4, Cohesin as regulator of R loop formation.
Cohesin-mutant cells accumulate DNA damage and have deficient DNA damage repair pathways and we have previously described the contribution of replication fork stalling to this phenotype (Tothova et al, JCI Insight 2021). Ongoing work in the lab is focused to understand the contribution of transcriptional stress in the form of R loop formation, and our preliminary data suggest that the cohesin complex is an important regulator of R loop formation.
5, Cohesin mutations as progression lesions in RUNX1 familial platelet disorder
Cohesin mutations are important progression lesions in a number of familial MDS/AML predisposition syndromes, including the RUNX1 familial platelet disorder (FPD), Trisomy 21 Down syndrome-associated acute megakaryoblastic leukemia (AMKL) and GATA 2 deficiency syndrome. We are interested in understanding the mechanisms by which cohesin mutations are selected for during MDS progression in these germline disorders and identifying ways to intercept progression to MDS and AML.
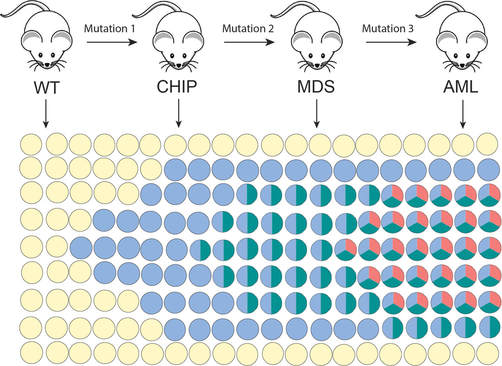
Photo credits: Francisco Marty, MD and Rebecca Gorelov, BA
|
|

|
|
|
|